Resident’s Forum
Section Editor: Jerry Tan, MD
Resident Section Editors: Sanjay Bhambri, DO, and Joshua Zeichner, MD
by Joseph F. Sobanko, MD; Lindsay Meijer, BS;
Thomas P. Nigra, MD
Dr. Sobanko is Chief Resident, Dermatology, Georgetown University Hospital/Washington Hospital Center, Washington, DC; Dr. Nigra is Chairman, Department of Dermatology, Washington Hospital Center, Washington, DC; and Ms. Meijer is a medical student at Georgetown University School of Medicine (MSIV), Washington, DC. The authors report no conflicts of interest. Section Editor: Jerry Tan, MD, FRCPC, is Adjunct Professor, University of Western Ontario, London, Ontario; President, Windsor Clinical Research Inc., Windsor, Ontario; and Consultant, Windsor Regional Hospital, Windsor, Ontario, Canada. He is also in private practice.
Abstract
Epithelioid sarcoma is a rare, high-grade, soft tissue tumor that has a known propensity for local recurrence, regional lymph node involvement, and distant metastases. We review the clinical and histological presentations of epithelioid sarcoma. Because epithelioid sarcoma presents innocuously, it is often mistaken as a benign process, which can result in insufficient treatment. Therefore, we emphasize the need for clinicians to consider this diagnosis when a slow-growing tumor is found on the distal extremity of a young male as the malignancy inherently portends a poor prognosis. Prognostic factors, such as local recurrence, regional metastatic disease, and tumor width, are discussed along with current treatment modalities, which include radical excision, sentinel lymph node biopsy, and radiation.
Introduction
Epithelioid sarcoma (ES) is a rare, high-grade malignancy that represents the most common primary soft tissue sarcoma of the hand. It was first described by Laskowski in 1961 as “sarcoma aponeuroticum” because of its involvement of aponeuroses and surrounding structures.[1] Less than a decade later, Enzinger coined the current term “epithelioid sarcoma” when recategorizing 62 previously misdiagnosed tumors.[2] Because of its epithelial and mesenchymal differentiation, this tumor was often mistaken for chronic inflammatory processes, necrotizing granulomas, and various fibrohistiocytic tumors.[3] These lesions were once documented clinically and histologically as malignant and nonmalignant conditions.[2] ES often presents in a banal fashion in young men, but its significance is great because of an inherent propensity for local recurrence in addition to regional lymph node involvement and distant metastases.[4–8] Misdiagnosis of this tumor can lead to delayed and improper treatment, adversely affecting patient survival.
Clinical Features
ES is characterized on physical examination as a firm, nontender, slow-growing tumor with a predilection for the hands, fingers, and forearms.[4,8–10] ES initially appears as a single nodule. However, at the time of diagnosis, multiple nodules representing local disease spread may be present.[7] The most common gross appearance is tan-white, nonencapsulated nodules with infiltrating borders.[2] Trauma has been noted to precede the tumor growth.[2] ES predominantly affects young adults in their second or third decade of life, but may occur at any age. Males are disproportionately affected with a ratio approaching 2:1.[2,5,8–13]
Because of the tendency toward nonpainful, indolent growth, ES may be present for months to years before the patient seeks medical attention.[2,5–14] This “benign nature” should not preclude the clinician from performing a biopsy if other clinical factors point toward malignancy. Occasionally, pain and tenderness, drainage, contractures, muscular weakness, numbness, and tingling may be experienced.[5,9,11,12] Limitation of function is usually not observed.
ES may be multifocal by the time a patient seeks medical care. The tumor may spread proximally along tendons, fascial planes, and aponeuroses, subsequently forming a nodular or pseudogranulomatous growth. The secondary nodules may be solitary or multiple, and range from a few millimeters to several centimeters in size.[2,4,5,8,9,12,14,15] They often undergo central necrosis, hemorrhage, and ulceration (Figure 1).[2,4,5,8,9,11,12,14,16] Periosteal bone invasion can occur.[2,14 ]Other locations that the tumor has been reported include the scalp, orbit, parotid gland, palate, penis, perineum, vulva, and buttocks.[17–22]
{kind=link}
Dissemination via the subdermal lymphatic vessels or the blood stream is a common feature of ES. The tumor can involve one or more lymph nodes when distant metastasis occurs, and these diseased nodes can enlarge up to several centimeters.[2,5,8,9,11,12,14] While adult patients with ES have demonstrated a higher rate of lymph node involvement than other soft tissue tumors, this trend is less evident in pediatric patients.[13] The lungs are the most common site of distant organ metastasis.[2,5,7,9]
Diagnosis
The diagnosis of ES is made via histopathological examination. Prior to obtaining a biopsy of a subcutaneous tumor, some clinicians may prefer to obtain imaging in order to assess for subclinical involvement and the best means of biopsy. Because of its ability to visualize soft tissue detail, magnetic resonance imaging (MRI), as opposed to computed tomography scans and X-rays, is the preferred modality for imaging.10 Following excision, MRI may also be used to differentiate residual tumor from postoperative disease.[10,23]
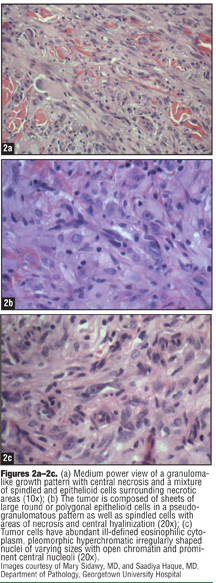
The three variants of ES are epithelioid, spindled, and mixed, with the principal form being epithelioid. These cells are large, round, oval, or polygonal, with abundant, deeply acidophilic cytoplasm and a clear or vesicular, centrally placed nucleus (Figures 2a–2c).[2,5,12] The cells contain well-defined cytoplasm and distinctive cell borders. Mitotic activity is almost always identified.[2,5,9,11,16] Ulceration may occur when ES originates at the superficial dermis and subcutaneous tissue. When ES derives from under the fascia it may become affixed to tendons, tendon sheaths, or fascia.[4,5,8,11] ES located on proximal portions of the body tends to be larger and deeper than the superficial forms.[9,12] Perineural and vascular invasion can also be observed with ES.[11]
{kind=link}
The proportion of spindle cells to epithelioid cells varies from tumor to tumor, and these cells may be arranged in a whorled pattern. Because of this variation, the histological appearance of ES has been mistaken for numerous benign and malignant entities (Table 1).[2,5,9,11–14] Evaluation of ES can also be difficult because some epithelioid cells aggregate into large nodules that may demonstrate central degeneration, necrotic debris, hyalinized collagen, or calcification. A heavy, lymphocytic, inflammatory infiltrate often surrounds the tumor cells.[5] Elucidation of the diagnosis is aided by consistently positive staining with the immunoreactants vimentin, epithelial membrane antigen, and cytokeratin.[24,25] Other stains, such as CD34, may also be positive (Table 2).
{kind=link}
{kind=link}
Prognosis
The course of ES is often unpredictable, and it is common for patients to present with extensive disease, lymph node metastases, or distant metastases. Enzinger’s initial report on ES demonstrated a local recurrence rate of 85 percent and a distant metastatic rate of 30 percent.[2] More recent authors report a metastatic rate approaching 50 percent.[5,8,9,11,26] As is true for most soft tissue sarcomas, the lungs are the principal site of metastatic disease.[2,5,7–9,10] ES has also been documented to metastasize to lymph nodes, skin, scalp, brain, digestive tract, liver, kidneys, and musculoskeletal system.[11]
Local recurrence often occurs within 1 to 2 years of treatment, and these patients often proceed to develop distant metastases.[5,8] Spillane et al found that both local recurrence and regional nodal metastases resulted in increased distant metastatic disease, thus decreasing overall survival.[8] The median postmetastatic survival was reported to be eight months.[8] Chase and Enzinger reported that the width of the tumor was directly proportional to the rate of metastasis and this directly correlated with a lower 10-year survival.[9] Numerous other case series have demonstrated the same findings.[5,7,9,10,12–14,16] Interestingly, female gender has been linked to a positive prognosis, but patient age at the time of diagnosis does not seem to alter survival.[5,7,9,10,12,14,16] Larger tumor depth, vascular invasion, and necrosis have been inconsistently reported to designate a poorer prognosis.[5,6,9,11,12,16]
Anatomic location of ES appears to play a role in prognostication. The overall survival and metastases-free survival are worse in lesions proximal to the elbow or knee.[7,15] This may be explained by the fact that “proximal-type” epithelioid sarcomas tend to present with unfavorable features, such as increased width, deep-seated tumor location, and preferential involvement of the pelvic, perineal, and genital regions.[7,13] Conversely, location on the distal extremities predicts a more favorable outcome.[13]
Management
Radical tumor excision is the primary treatment for patients with ES. Negative tumor margins must be obtained, but optimal function, particularly of the upper limb, remains a potential goal.[2,5,8–11,13,16] Marginal excision is inadequate due to the high rate of local recurrence and distant metastasis.[5] Routine sentinel lymph node biopsy (SLNB) does not appear to be supported by data, though it is sometimes advocated due to the high rate of regional recurrence. SLNB is also thought to identify individuals who may benefit from systemic therapy.[7,10] Therapeutic lymph node dissection is indicated when lymph node metastases are present.[6,8,11,14] Halling et al suggest that, in the case of in-transit metastases, surgical resection of single or a small number of metastatic lesions may result in increased long-term survival.[5] Local recurrence may necessitate amputation.[14]
While the efficacy of radiation therapy as an adjunct to surgery has been clearly demonstrated in soft tissue sarcomas, studies of ES are limited due to small sample size and limited follow up.[27,28] A decreased risk of local recurrence has been reported with the addition of preoperative or postoperative local radiation therapy when combined with radical surgery.[4,5,7,8] Radiation can also be considered for patients with marginal primary resection, local regional recurrence, or palliative treatment.[8] Adjuvant chemotherapy appears to be indicated in the case of metastatic disease, but this is less clear in nonmetastatic ES.[26] Additional studies with long-term follow up would be helpful to further address the role of adjuvant therapy in the treatment of ES. Limited experience with isolated limb perfusion indicates that it may be beneficial for multifocal or large unresectable tumors, allowing for reduction of tumor size with subsequent radical excision.[7,8]
Conclusion
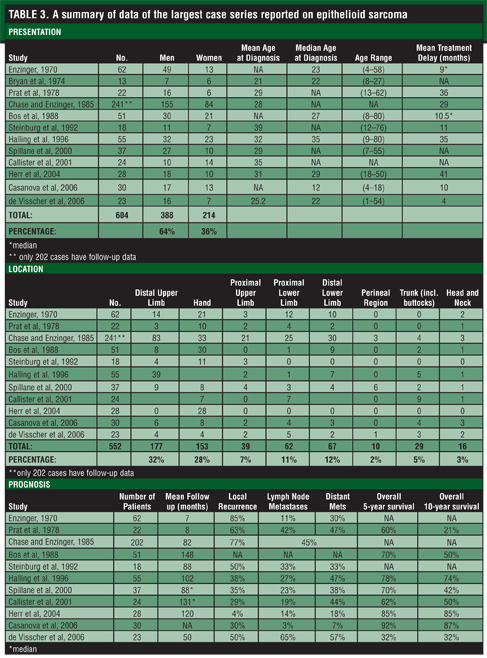
ES is a rare, high-grade, soft tissue tumor that has a known propensity for local recurrence, regional lymph node involvement, and distant metastases. This malignancy can easily be mistaken for a benign process due to its often innocuous presentation. Because of its potential for aggressive behavior, clinicians must be aware of the presenting behavior of ES in order to avoid misdiagnosis. Unusual nodules, particularly on the distal extremity of young men, must be closely scrutinized if there is a suspicion of malignancy. Aggressive treatment with preoperative or postoperative radiation therapy combined with wide local excision appears to be indicated once diagnosis is established via histopathological confirmation[.5,7] SLNB and lymphadenectomy may also be helpful in certain subsets of patients.[7,12] Close, long-term follow up is necessary because recurrence and metastases may occur long after definitive treatment. Table 3
summarizes the data reported in the largest case series of ES over the past four decades.[1,5–10,12–14,16]
{kind=link}
References
1. Laskowski J. Sarcoma aponeuroticum. Nowotory. 1961;11:61–67.
2. Enzinger FM. Epithelioid sarcoma. A sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029–1041.
3. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;265–275.
4. Schimm AJ, Suit HD. Radiation therapy of epithelioid sarcoma. Cancer. 1983;52:1022–1025.
5. Halling AC, Wollan PC, Pritchard DJ, Vlasak R, Nascimento AG. Epithelioid sarcoma: a clinicopathologic review of 55 cases. Mayo Clin Proc. 1996;71:636–642.
6. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft-tissue sarcoma. Ann Surg Oncol. 2000;7:218–225.
7. Callister MD, Ballo MT, Pisters PW, et al. Epithelioid sarcoma: results of conservative surgery and radiotherapy. Int J Radiat Oncol Biol Phys. 2001;51:384–391.
8. de Visscher SAHJ, van Ginkel RJ, Wobbes T, et al. Epithelioid sarcoma: still an only surgically curable disease. Cancer. 2006;107:606–612.
9. Chase DR, Enzinger FM. Epithelioid sarcoma. Diagnosis, prognostic indicators, and treatment. Am J Surg Path. 1985;9:241–263.
10. Herr MJ, Harmsen WS, Amadio PC, Scully SP. Epithelioid sarcoma of the hand. Clin Orthop Relat Res. 2004;431:193–200.
11. Woodruff JM, Marcove RC. Epithelioid sarcoma: an analysis of 22 cases indicating the prognostic significance of vascular invasion and regional lymph node metastasis. Cancer. 1978; 41:1472–1487.
12. Bos GD, Pritchard DJ, Reiman HM, et al. Epithelioid sarcoma. An analysis of fifty-one cases. J Bone Joint Surg Am. 1988;70:862–870.
13. Cassanova M, Ferrari A, Collini P, et al. Epithelioid sarcoma in children and adolescents. A report from the Italian Soft Tissue Committee. Am Cancer Society. 2006;106:708–717.
14. Bryan RS, Soule EH, Dobyns JH, Pritchard DJ, Linscheid RL. Primary epithelioid sarcoma of the hand and forearm. A review of thirteen cases. J Bone Joint Surg. 1974;56A:458–465.
15. Guillou L, Wadden C, Coindre JM, Krausz T, Fletcher CD. “Proximal-type” epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol. 1997;4:491–495.
16. Steinberg BD, Gelberman RH, Mankin HJ, Rosenberg AE. Epithelioid sarcoma in the upper extremity. J Bone Joint Surg Am. 1992;74:28–35.
17. Jameson CF, Simpsom MT, Towers JF. Primary epithelioid sarcoma of the hard palate: A case report. Int J Oral Maxillofac Surg. 1990;19:240.
18. Huang DJ, Stanisic TH, Hansen KK. Epithelioid sarcoma of the penis. J Urol. 1992;147:1370.
19. Aartsen EJ, Albus Lutter CE. Vulva sarcoma: clinical implications. Eur J Obstet Gynecol Reprod Biol. 1994;56:181.
20. White VA, Heathcote JG, Hurwitz JJ, et al. Epitheliod sarcoma of the orbit. Opthamology. 1994;101:1680.
21. Johannssen V, Janig U, Werner JA. Epithelioid sarcoma of the parotid region (German). Laryngorhinootologie. 1996;75:556.
22. Suwantemee C. Primary epithelioid sarcoma of the scalp. Am Soc of Plastic Surg. 1999;104(3):785–788.
23. Hanna SL, Kaste S, Jenkins JJ, et al. Epithelioid sarcoma: clinical, MR imaging and pathologic findings. Skeletal Radiol. 2002;31:400–412.
24. Daimaru Y, Hashimoto H, Tsuneyoshi M, Enjoji M. Epithelial Profile of epithelioid sarcoma: an immunohistochemical analysis of eight cases. Cancer. 1987;59:134.
25. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934–942.
26. Ross HM, Lewis JJ, Woodruff JM, Brennan MF. Epithelioid sarcoma: clinical behavior and prognostic factors of survival. Ann Surg Oncol. 1997;4:491–495.
27. Lindberg R, Martin R, Romsdahl M, Barkley H. Conservative surgery and postoperative radiotherapy in 300 adults with soft tissue sarcomas. Cancer. 1975;35:1478–1483.
28. Russell W, Cohen J, Edmondson J, et al. Staging system for soft tissue sarcoma. Semin Oncol. 1981;8:156–159.